








Rediscovering Biology: Molecular to Global Perspectives
Online Textbook and Video
This online textbook chapter supports and extends the content of the Evolution and Phylogenetics video. The chapter covers the evolution of phylogenetics as a field of study.
1. Introduction
“Systems of classification are not hat racks, objectively presented to us by nature. They are dynamic theories developed by us to express particular views about the history of organisms. Evolution has provided a set of unique species ordered by differing degrees of genealogical relationship. Taxonomy, the search for this natural order, is the fundamental science of history.”
– Stephen J Gould1
Introduction
Perhaps the most striking feature of life is its enormous diversity. There are more than one million described species of animals and plants, with many millions still left undescribed. (See the Biodiversity unit.) Aside from its sheer numerical diversity, organisms differ widely and along numerous dimensions – including morphological appearance, feeding habits, mating behaviors, and physiologies. In recent decades, scientists have also added molecular genetic differences to this list. Some groups of organisms are clearly more similar to some groups than to others. For instance, mallard ducks are more similar to black ducks than either is to herons. At the same time, some groups are very similar along one dimension, yet strikingly different in other respects. Based solely on flying ability, one would group bats and birds together; however, in most other respects, bats and birds are very dissimilar. How do biologists organize and classify biodiversity?
In recent decades, methodological and technological advances have radically altered how biologists classify organisms and how they view the diversity of life. In addition, biologists are better able now to use classification schemes for diverse purposes, from examining how traits evolve to solving crimes. These advances have strengthened evolutionary biology as a theory: a theory in the scientific sense, meaning a “mature coherent body of interconnected statements, based on reasoning and evidence, that explains a variety of observations.”2 Molecular biology, genetics, development, behavior, epidemiology, ecology, conservation biology, and forensics are just a few of the many fields conceptually united by evolutionary theory.
2. A Brief History of Classification
Taxonomy, the practice of classifying biodiversity, has a venerable history. Although early natural historians did not recognize that the similarities and differences among organisms were consequences of evolutionary mechanisms, they still sought a means to organize biological diversity. In 1758 Carl Linn proposed a system that has dominated classification for centuries. Linnaeus gave each species two names, denoting genus and species (such as Homo sapiens). He then grouped genera into families, families into orders, orders into classes, classes into phyla, and phyla into kingdoms. Linn identified two kingdoms: Animalia (animals) and Plantae (plants). Biologists generally accepted the idea of evolution shortly after the publication of Darwin’s The Origin of Species and, since Linnaeus’ classification system, they have described an immense number of species. Despite these facts, taxonomy changed little until the 1960s.
The first major break from the Linnean model came from Thomas Whittaker. In 1969 Whittaker proposed a “five kingdom” system in which three kingdoms were added to the animals and plants: Monera (bacteria), Protista, and Fungi. Whittaker defined the kingdoms by a number of special characteristics. First, he specified whether the organisms possessed a true nucleus (eukaryotic) or not (prokaryotic). Because Monera are prokaryotic and virtually all are unicellar, they are distinct from the other four eukaryotic kingdoms. With few exceptions, the eukaryotic unicellular organisms were placed into the kingdom Protista.
The three multicellular eukaryotic kingdoms distinguish themselves by the general manner in which they acquire food. Plants are autotrophs and use photosynthetic systems to capture energy from sunlight. Animals are heterotrophs and acquire nutrients by ingesting plants or other animals, and then digesting those materials. Fungi are also heterotrophs but, unlike animals, they generally break down large organic molecules in their environment by secreting enzymes. Unicellular organisms use a variety of modes of nutrition. (See the Microbial Diversity unit.)
The five kingdoms system was certainly an advance over the previous system because it better captured the diversity of life. Three groups — bacteria, fungi, and protists – did not fit well into either the animal or plant category. Moreover, each of these three groups appeared to possess diversity comparable to that of animals or plants. Thus, the designation of each as a kingdom seemed fitting.
In the years since Whittaker’s system was developed, however, new evidence and new methods have shown that the five-kingdom system also fails to adequately capture what we now know about the diversity of life. Microbial biologists became aware of these limitations as they discovered unicellular organisms that appeared to be prokaryotic, but were extremely distinct in ultrastructure and other characteristics from the traditional bacteria. Some of these unusual prokaryotes lived in hot springs and other places where the temperatures were near, or even above, the boiling point of water (the thermophiles). Others, the extreme halophiles, were able to tolerate salt concentrations as high as five Molar, roughly ten times the concentration of seawater. (See the Microbial Diversity unit.) DNA sequence data also increasingly suggested that these prokaryotes were most unlike the traditional bacteria.

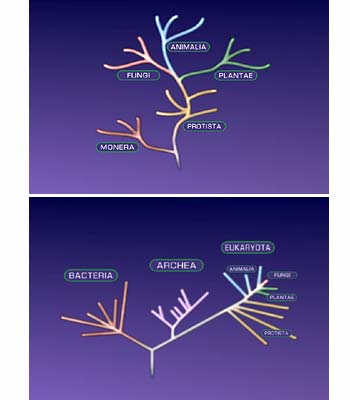
Figure 1. The older five-kingdom tree of life, which has been replaced by Woese’s three-domain tree.
The microbal evolutionist Carl Woese proposed a radical reorganization of the five kingdoms into three domains. (See the Microbial Diversity unit.) Starting in the 1980s Woese’s scheme has been increasingly accepted by evolutionary biologists and is now the standard paradigm. In his classification system, Woese placed all four eukaryotic kingdoms into a single domain called Eukarya, also known as the eukaryotes. He then split the former kingdom of Monera into the Eubacteria (bacteria) and the Archaea (archaebacteria) domains. Woese then placed most of the “unusual” prokarytes in the Archaea, leaving traditional bacteria in the Eubacteria. The Woese classification represents a demotion of the animals and plants as individual kingdoms. This is consistent with recent discoveries of more diversity among microbes than between animals and plants.
Unlike Whittaker’s five kingdoms system, Woese’s three domains system organizes biodiversity by evolutionary relationships. After a discussion of the methodology of contemporary evolutionary classification, we will examine the methods Woese used and the justification for his system.
3. Cladistics and Classification
Except for his last sentence where he used the word “evolved,” Charles Darwin never mentioned “evolution” in The Origin of Species. Instead, he used the phrase “descent with modification.” Evolutionary classification today is based on those two central features of evolution: groups of organisms descend from a common ancestor and, with the passage of time, acquire modifications.
Cladistic analysis, also known as cladistics and phylogenetic systematics, is the main approach of classification used in contemporary evolutionary biology. The German taxonomist Willi Hennig developed cladistics in 1950, but his work was not widely known until it was translated into English in 1966. After scientists began using molecular data in classification, Hennig’s cladistics became increasingly adopted.
Cladistic analysis starts with the assumption that evolution is a branching process: ancestral species split into descendant species, and these relationships can be represented much like family trees represent genealogies. The “trees” obtained by such analyses are called phylogenies. These phylogenies should be viewed as testable hypotheses, subject to either confirmation or rejection depending on new evidence. Of course, hypotheses differ as to how much support they have. Some are so well supported (such as that humans share a closer common ancestor to chimpanzees than either share with lemurs) that they are exceedingly unlikely to be overturned.
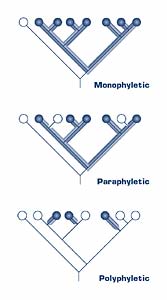
In cladistic analysis, groups of organisms, known as taxa, are arranged into clades that are then nested into larger clades. The term “taxa” (singular “taxon”) can be applied to groups of any size. Taxa that are each others’ closest relatives are called sister taxa. Each clade should be monophyletic; that is, all members share a single common ancestor, and all descendants of that ancestor are included in the clade. In contrast, a polyphyletic group is one in which the members are derived from more than one common ancestor. What if all of a particular clade’s members share a common ancestor but not all taxa that share that common ancestor are included in that group? Such a group is called paraphyletic.

Figure 2. Examples of monophyletic (top), paraphyletic (middle), and polyphyletic (bottom) trees.
Taxonomists following cladistic analysis place taxa into clades based on the derived character states that the taxa share. For example, a wing is a character. The presence or absence of a wing would be alternative character states. Other features of a wing (such as its shape and size, and how it develops) could also be character states. Aside from the presumption that characters are independent of one another, any trait can be a character. In principle, there is no difference between the analysis of morphological and molecular characters. The characters used most often in molecular phylogenies are the nucleotide positions of the examined DNA molecule(s); thus, the character states are the actual nucleotides at that position. Shared, derived characteristics are known as synapomorphies.
That taxonomists would classify taxa based on similarity makes sense. After all, like goes with like. But why would they consider only the derived shared character states? Why not consider all character states, including those that are primitive? The rationale is that the primitive characters do not reveal information about which groups share more recent common ancestors; the primitive character states would only contribute noise to the system. In classifying different groups of birds that all fly, whether they fly does not contribute information. In fact, in classifying flightless birds, considering the ancestral state (flighted) can actually distort the obtained phylogeny away from the true phylogeny. For these reasons, only synapomorphies (shared, derived character states) are considered in the analysis. In practice, taxonomists often have difficulty in distinguishing between which character states are primitive and which are derived.
For what reasons can taxa share synapomorphies? One possibility is that they share a common ancestor. This is called homology. While cladistic analysis assumes that most synapomorphies will arise by homology, they can arise by other ways. One possibility is convergence: different lineages that do not share a recent common ancestor evolve to the same character state. An obvious example is that both bats and birds have wings; however, these were independently derived, most likely owing to similar selective forces. This example is obvious because so many other characters place bats closer to non-winged clades (other mammals) than to birds. Yet, less obvious cases can be resolved only after cladistic analysis. Another possible reason why non-homologous character states can be similar is a reversal in which mutation or selection causes the derived character state to revert to the ancestral state.

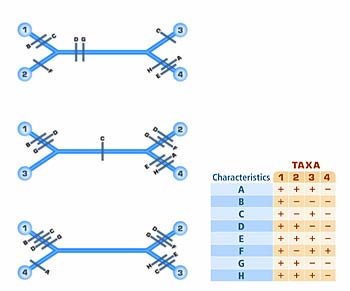
Figure 3. Three possible unrooted trees are shown. The top tree assumes nine changes in character state occurred (each change is represented by a mark), the middle tree assumes ten changes, and the bottom tree assumes eleven. Because the top tree assumes the fewest changes, it is the most parsimonious tree.
How does cladistic analysis work, especially given the possibility of conflicting data generated by reversals and convergence? Taxonomists, like scientists in general, start with the principle of parsimony — that the shortest, most simple, and direct path is most likely to be the correct one. In one commonly used method, parsimony analysis, the taxonomist searches for the most parsimonious tree; that is, the one that requires the fewest number of evolutionary transitions. Consider the example in Figure 3: three possible phylogenies exist. Based on the data given, for phylogeny (A) to occur, we must postulate a total of x evolutionary changes. Phylogeny (B) requires postulating y changes and phylogeny (C) requires postulating z changes. Because (B) requires the fewest changes, it is the most parsimonious tree.

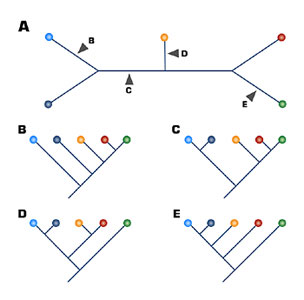
Figure 4. Panel A shows an unrooted tree. Panels B, C, D, and E should be the resulting rooted trees, when root is placed in each of the corresponding positions.
The most parsimonious tree may not necessarily represent the true phylogenetic relationships. Perhaps certain types of transitions are more likely or evolved more easily than are others. It is often difficult to know before doing the analysis, which changes are most likely. Thus, taxonomists generally resort to the fallback position that all changes are equally likely. There are some cases, particularly with molecular data, where there is good prior knowledge of variation in the likelihood of different changes. For instance, certain types of mutations are more likely than others are. Transitions (changes from a purine — A or G — to the other purine, or a pyrimidine — C or T — to the other pyrimidine) are more likely than transversions (changes from a purine to a pyrimidine or vice-versa). Using increasingly statistical techniques, such as maximum likelihood analysis, taxonomists can adjust for these situations.
Figure 4 shows an example of an unrooted tree. Unrooted trees do not display the directionality of evolution, only patterns of relatedness. A unrooted tree can be rooted, but for any given unrooted tree there are many possible rooted trees that can be derived. Rooting a tree usually requires identification and use of an outgroup — a taxon that is more distantly related than the taxa contained within the tree. For instance, given an unrooted tree containing the great apes (humans, chimpanzees, gorillas, orangutans, and gibbons), one could use a species of monkeys, such as baboons, as an outgroup. (See the Human Evolution unit.) In practice, taxonomists often use multiple outgroups to refine the analyses.
4. Applications of Molecular Phylogenetics
Although the methods used in cladistic analysis are the same for both molecular and morphological characters, molecular data provides several advantages. First, molecular data offers a large and essentially limitless set of characters. Each nucleotide position, in theory, can be considered a character and assumed independent. The DNA of any given organism has millions to billions of nucleotide positions. In addition, the large size of the genome makes it unlikely that natural selection will be strongly driving changes at any particular nucleotide. Instead, most nucleotide changes are “unseen” by natural selection, subject only to mutation and random genetic drift. If we were to assume that the driving force of natural selection is less prevalent for molecular characters, then we should assume that the probability of convergence for molecular characters is also.
By selecting a particular class of morphological characters, researchers may also bias the analysis in such a way that groups with certain characteristics cluster with others for reasons other than homology. For instance, if the set of characters were weighted toward those involved in carnivory, carnivorous animals may cluster together — not because of homology but because of shared function. This problem would be less likely if using molecular characters.
Another advantage of molecular data is that all known life is based on nucleic acids; thus, studies involving any type of taxa can use DNA sequence data. Some genes or regions of genes evolve quickly. These are most useful in studies of closely related taxa. Conversely, other genes (or regions) are slower to evolve; these are the most useful for studies of more distantly related organisms. At the extreme, some evolutionarily related genes have been found in disparate organisms such as yeast and humans. Rates by which sections of DNA evolve are primarily determined by the extent of functional constraint. Genes and positions within genes that are the most useful generally are the slowest to evolve. This is because they are the least able to tolerate mutational change without substantially reducing the fitness of the individuals that harbor them. Many of these very conserved genes play a role in development. (See the Genetics of Development unit.)
Starting in the late 1970s Carl Woese took on an ambitious project – determining the relationships of all life, which resulted in the reorganization of the tree of life. To do this, Woese and his associates took advantage of a molecule that evolves extremely slowly — rDNA, the DNA that encodes a small subunit of ribosomal RNA. They found that the sequences cluster in three groups corresponding to the eukaryotes (Eukarya), the archaea, and the eubacteria. We discussed these three domains earlier.
The three-domains model was controversial for several reasons. First, the conclusions Woese drew were initially based on evidence from a single gene. Perhaps there was something unusual about the way that small subunit of rDNA evolved, his critics said. That controversy was easily solved by generating more data. Sequences from other genes that evolve slowly seemed to confirm the rationale for the three domains. A more fundamental problem was that Woese’s tree was unrooted. If each domain represents a monophyletic group, three possiblilties existed: (1) that the eubacteria and archaea are sister groups, with the eukaryotes branching off first; (2) that eubacteria and eukaryotes are sister groups; or (3), that archaea and eukaryotes are sister groups. Woese himself suspected this third possibility. A fourth possibility was that the root of the tree lied within one of the domains and, therefore, the domain was not monophyletic. To root a tree, one generally requires an outgroup. But what is the outgroup to all known life? Rocks?
Margaret Dayhoff proposed an ingenious solution to this rooting dilemma: using ancestral genes that are present in multiple copies in the same organism because of gene duplication. If there were such genes that had duplicated before the split among the three domains, these could be used as outgroups to root the tree of life. In 1989, many years after Dayhoff’s suggestion, Naoyuki Iwabe and colleagues used this approach.3 Organisms in all three domains have two distinct genes that code for the two subunits (alpha and beta) of the enzyme that hydrolyzes ATP to yield energy, ATPase. DNA sequence similarity strongly suggests that these two genes are derived from a gene duplication pre-dating the divergence of the domains. The ATPase-alpha tree, using an ATPase-beta gene as an outgroup, showed that each of the domains was monophyletic, and that eukaryotes and archaea are sister groups. The same result was obtained when ATPase-beta was used as an outgroup to root the ATPase-alpha tree. Similar trees were obtained with other pairs of duplicated genes. In conclusion, Woese was right.
5. HIV and Forensic Uses of Phylogenetics
Phylogenetic methods have been used to solve practical problems, including determining the sources of infection from HIV. This retrovirus evolves at an extremely rapid rate, owing to its exceptionally high mutation rate. In fact, sequences of HIV genes taken from the same infected individual can be as different as sequences from some homologous genes in humans and birds. Its rapidity of evolution works to HIV’s advantage as it wreaks havoc on the immune system. On the other hand, scientists can take advantage of that rapid evolution to study the relationships between HIV and other similar viruses.
Researchers at the Centers for Disease Control and Prevention (CDC) used phylogenetic systematics of HIV for forensic purposes. During the early 1990s a Florida dentist was suspected of transmitting HIV to several of his patients. After the first case of probable transmission surfaced, the dentist wrote an open letter to his patients suggesting that they be tested for HIV. At least ten of the patients tested positive for HIV. However, a few of the infected individuals had other risk factors; therefore, there was the distinct possibility that they had not been infected by the dentist. The CDC researchers sequenced the HIV gp120 gene from several viral isolates taken from the dentist, his infected patients, and non-patients who were also infected. From the phylogeny constructed based on the HIV sequence data, they first denoted what they called the “dentist clade.” This monophyletic group contained sequences from the HIV sequences collected from the dentist but not from the non-patients. Five of the patients had viral sequences that were contained in the dentist clade. These patients also lacked other risk factors. Thus, by strong inference, the CDC researchers determined that the dentist had infected these five patients.
There was some controversy over whether or not the dentist clade identified in the CDC study was reliable. Nucleotides in the HIV gp120 gene do not evolve in same way as in other genes. Instead of transitions being universally more prevalent than transversions, as is the case in most genes, A to C transversions are more frequent than transitions of C to T. There was also concern about the types of algorithms used. To address these concerns, David Hillis, John Huelsenbeck, and Cliff Cunningham re-analyzed the data of the CDC study. They found that, under nearly all circumstances, the same dental clade was obtained.4 Thus, the results were statistically reliable. Investigators are using similar studies to determine the source of the anthrax used in the attacks of October 2001.
6. The Origin of Bats and Flight

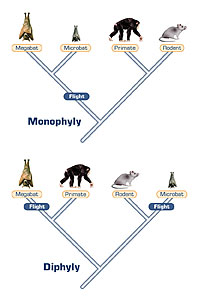
Figure 5. Alternative possibilities of bat phylogeny. Top: Bats form a monophyletic clade, in which flight evolved once in mammals. Alternately, bottom, bats are diphyletic, and flight evolved twice in mammals.
Molecular phylogenetics are often most useful when there is conflict among the phylogenies constructed with different morphological character data sets. For instance, molecular data have helped settle the question of whether bats are a monophyletic group – that is, whether they share a common ancestor not shared by non-bats. In the 1980s several morphological analyses challenged the traditional view that bats (order Chiroptera) were monophyletic. The studies proposed that the large fruit-eating Megachiroptera (megabats) were actually more closely related to primates than they were to the smaller insect-eating Microchiroptera (microbats). The studies based the megabat-primate grouping on synapomorphies that included features of the penis, brain, and limbs. The implication of this reclassification was that flight evolved more than once within mammals.

Figure 5a. Photograph of an example of a megabat, the African fruit bat.
Courtesy of Transvaal Museum, South Africa.
Spurred by this controversy, several research groups performed cladistic analyses of bats using molecular data during the early 1990s. For example, Loren Ammerman and David Hillis sequenced mitochondrial DNA sequences from many mammals, including two species of microbats, two species of megabats, a tree shrew, a primate, and several outgroups. From their data, the most parsimonious tree that assumed bat monophyly was ten steps shorter than the most parsimonious tree that assumed bats were not monophyletic. Statistical analysis showed that bat monophyly was significantly more parsimonious than the absence of bat monophyly. Other molecular phylogenetic studies, using a variety of different classes of genes, showed the same pattern of bat monophyly. These researchers also indicated that convergence is the most likely reason why some derived morphological character states seem to be shared by primates and bats.5

Figure 5b. Photograph of an example of a microbat, the Mexican freetail bat.
Courtesy of Robert Bloomberg.
Other researchers raised the objection that these early molecular phylogenetic studies did not take into account biases in the way that sequences evolve. Specifically, the critics noted that both microbats and macrobats have DNA with a higher proportion of G’s and C’s than A’s and T’s. It is well known that organisms that have higher metabolic rates will have higher G-C content. Thus, the critics argued, perhaps the apparent monophyly of bats that was observed in the molecular studies is due to convergent evolution toward high G-C content and not homology. Using various methods, subsequent molecular phylogenetic studies took the bias in nucleotide changes into account. One simple method was to split the DNA sequences into A-T rich and G-C rich regions and do a separate analysis on each. Even after nucleotide sequence bias was discounted, the most parsimonious phylogenies still showed that all bats had a single common ancestor. This support for bats as a monophyletic group is also strong evidence for flight evolving only once in mammals.
The monophyly of bats is an example where molecular data shored up the traditional phylogeny against challenges posed by some morphological characters. In contrast, there are also occasions where analysis of the molecular data provided an unexpected answer. One such example is the example of the evolutionary history of whales, which is discussed in detail in the video.
7. Challenges
There have been tremendous advances in comparative evolution brought on by the new methods of phylogenetic analysis and burgeoning amounts of DNA sequence data; however, the field is not without challenges and limitations. Some of these challenges are due to features of the organism and some are due to limitations of the tools we currently possess.
One feature of the organism that presents a challenge is the horizontal transfer of genes across different species. In the standard mode of vertical transmission, genes are transmitted from parent to offspring (whether by sexual or asexual means). Genetic material can also be exchanged among different organisms, especially bacteria. This general type of transmission is called lateral gene transfer. One mode by which lateral gene transfer can occur is conjugation, whereby some bacteria exchange genes (plasmids or small parts of the bacterial chromosome) by physical contact. Bacteriophages can also mediate lateral gene transfer by cross-infection. Amazingly, these processes that result in lateral gene transfer can occur among bacteria that differ by as much as fifteen percent at the DNA sequence level. The implication of widespread and random lateral transfer of genes is that the genetic structure of bacteria can be mosaic – different genes or gene regions may have different histories. If lateral transfer is sufficiently pervasive, it could lead to the inability of constructing the true phylogeny for all bacteria. (See the Microbial Diversity unit.)
The most dramatic case of lateral gene transfer involving eukaryotes is the endosymbiotic origin of mitochondria. This view, championed by Lynn Margulis, speculates that these ATP-producing organelles were once free-living prokaryotes that were engulfed by a proto-eukaryote — an idea now strongly supported. The evidence includes similarities of ribosomal structure, sensitivity to antibiotics, and DNA sequences between mitochondria and prokaryotes. The major controversy is when and how this process occurred. Other eukaryotic organelles have been shown to probably have endosymbiotic origins. The conventional wisdom, however, is that lateral gene transfer involving eukaryotes was limited from these exceeding rare endosymbioic events.

Figure 6. Proposed by W. Ford Doolittle, this view of early evolution suggests multiple primitive cells as ancestors to the three domains, and illustrates lateral gene transfer among early organisms.
Recent evidence strongly suggests that lateral gene transfer involving eukaryotes may be more prevalent than once thought. In some DNA sequences, bacterial or archaeal sequences cluster in clades that are otherwise strictly eukaryotic. The extent to which lateral gene transfer among the kingdoms and within the eukaryotes has occurred is still a matter of controversy and inquiry. The implications for our ability to construct accurate phylogenies for these “deep” relationships are also controversial. There appears to be a continuum of the degree to which different genes transfer across distantly related taxa. Some researchers have argued that we may be able to get around the problem of lateral gene transfer by choosing genes that display very little — if any — horizontal gene transfer.
Another major challenge to comparative evolution is that the methodology of phylogenetic systematics is computationally extensive. The number of potential trees increases extremely quickly — faster than exponentially — as the number of taxa increases. For three taxa, there are only three possible rooted trees. For a given data set, one can readily determine by inspection which tree is the most parsimonious. Given seven taxa, it would be exceedingly painstaking for a person to search for the most parsimonious tree through the 10,395 rooted possibilities; however, a desktop computer with the correct software could search among all of these possibilities in a tiny fraction of a second.
Increasing computing power alone will not solve this problem. At twenty taxa, the number of possible rooted trees exceeds 8 times 1021 – a number of similar magnitude to the total number of cells in all living human beings. Soon after this point, it becomes impractical for computers to search through all the possibilities to find the most parsimonious one. Given fifty taxa, it would take literally longer than the age of the universe to search through every single possible unrooted tree — even if computers were a million times faster than they are now. Therefore, phylogenetic systematics must employ methods other than searching every single possible tree when evaluating data sets that involve a large number of taxa. One method is to collapse taxa that are known (by other information) to be close relatives into a single taxon to make the analysis more feasible. Researchers have also used various searching approaches, sometimes called heuristics. This approach uses algorithms to identify regions of “tree space” that are likely to contain very parsimonious trees. These heuristic methods may not always identify the best tree, but they will identify trees that are nearly as parsimonious as the best tree most of the time.
8. Coda: The Renaissance of Comparative Biology
We are witnessing a renewal of interest in comparative approaches to studying function. Biology in the 1800s was almost entirely comparative. In the twentieth century we moved into a strongly reductionistic period of genetics, developmental biology, and physiology. This trend only intensified with the rise of molecular biology, particularly after the elucidation of the structure of DNA in 1953. At that time, comparative biology was marginalized as just “natural history.” At the turn of the twenty-first century comparative approaches have staged a strong comeback. In large part, this renaissance is due to the revolution in data gathering (particularly of DNA sequences) and the effort already devoted to establishing particular model systems. In contrast to the comparative biology of ninteenth century, today’s comparative evolutionary biology rests on a strong foundation of functional genetics.
9. End Notes
-
- Gould, S. J. 1987. Natural History.
- Futuyma, D. J. 1998. Evolutionary Biology. 3d ed. Sunderland MA: Sinauer Press, p. 11.
- Iwabe, N., K. Kuma, M. Hasegawa, S. Osawa, and T. Miyata. 1989. Evolutionary relationship of the archebacteria, eubacteria, and eukaryotes inferred from phylogenetic trees of duplicated genes. Proceedings of the National Academy of Sciences 86:9355-9359.
- Hillis, D. M., J. P. Huelsenbeck, and C. W. Cunningham. 1994. Application and accuracy of molecular phylogenies. Science264:671-77.
- Ammerman, L. K., and D. M. Hillis. 1992. A molecular test of bat relationships:Monophyly or diphyly? Systematic Biology41: 222-32.