



Rediscovering Biology: Molecular to Global Perspectives
Emerging Infectious Diseases Expert Interview Transcript: Capt. Daniel Carucci, M.D., PhD
 Director, NMRC Malaria Program
Director, NMRC Malaria Program
Dr. Carucci is the Director of the Malaria Program at the Naval Medical Research Center where he oversees Navy research efforts on malaria, focusing on vaccine development and novel approaches to protecting individuals from the disease. He is also the Department of Defense coordinator for the Malaria Genome Project, where he oversees all DoD sponsored research in malaria genomics and proteomics.
Interview Transcript
Dr. Carucci is the Director of the Malaria Program at the Naval Medical Research Center where he oversees Navy research efforts on malaria, focusing on vaccine development and novel approaches to protecting individuals from the disease. He is also the Department of Defense coordinator for the Malaria Genome Project, where he oversees all DoD sponsored research in malaria genomics and proteomics.
Although malaria is an ancient disease, is it still a major health problem?
It is a problem. There are more people infected from malaria in the world today than ever have been infected in history. It is interesting: you talk to people who really are not apprised of what is going on with people’s health in a lot of the developing world, and people don’t have a good appreciation, I don’t think, for the amount of infectious diseases that are out there infecting people. HIV obviously is in the news a lot, but the developing world is being devastated by infectious diseases, and it’s not only impacting the health of the indigenous people, but it’s an economic problem as well, because it reduces productivity, a country’s productivity is reduced and its wealth is reduced, and everyone suffers because their population is malarial.
Malaria is probably second only to tuberculosis in number of people that actually die from it every year. There are about 300 to 500 million cases of malaria each year. There are about 3 million deaths every year. Most of those are in children in sub-Saharan Africa, where the malaria represents about 10% of childhood mortality. About a million children die every year in sub-Saharan Africa, and I think actually those estimates are probably an underestimate of what’s going on.
The people who die from malaria are kids, really. Malaria has been associated with people. It has co-evolved with people. It’s a parasite. It has learned to adapt to a very, very violent and hostile immune system inside the human body, and it has adapted. It’s a very smart organism, and we’re going to talk a little bit about the malaria genome and how we hope that, by understanding more about the parasite, we’ll be able to develop better interventions, both drugs and vaccines, through that understanding.
If one gets a vaccine shot, won’t they be fine?
That’s the kind of thing that people often ask about. People know that I’m involved in tropical medicine, and they say, “Listen, I’m going to Africa. Can I just get that shot for malaria prevention?” And I tell them, “Well, no, there is no shot for malaria. I mean you can take pills. These are drugs that you can take to prevent you from getting malaria, and those drugs generally work pretty well. The most important drug that we had developed over the past 50 years is a drug called Chloroquine. And because of its safety, its low cost and how effective it was, the World Health Organization, in the late 1940s and early 1950s, embarked on a malaria eradication program, and that combined the drug with DDT spraying to eradicate the mosquito.
In the late 1950s, the World Health Organization embarked on a malaria eradication program and that really was conceived because of two things. One was the development of Chloroquine, which is a very safe and effective and inexpensive drug to treat malaria in those individuals who are infected, and prevent it from getting to other people. The second was the development of the use of DDT as an insecticide against the mosquito vector. So the combination of those two things, the use of Chloroquine to clear the people of parasite infections in their blood, and the use of DDT as an insecticide, meant that we had what we thought were the tools available to eradicate malaria. It was a two-pronged strategy to eradicate the disease.
Unfortunately, in the early 1960s, two areas of resistance developed. One was in the Venezuela-Colombia area in South America, and the other one was in Southeast Asia. And these are areas where drug resistance developed-the parasite developed resistance against Chloroquine, and this resistance spread relatively very quickly throughout those areas of the world, sparing Africa until about 1978.
Now Africa is the hotbed of malaria. This is where malaria impacts most greatly on people, and so Chloroquine resistance, when it appeared on the East Coast of sub-Saharan Africa in 1978, really was an ominous sign, because now suddenly Chloroquine resistance was going to spread throughout Africa. Now, if you took a map of where the malaria parasite exists and where Chloroquine resistance exists, they superimpose each other completely, so there is Chloroquine resistance everywhere that there is malaria. We do have newer drugs. The other drugs that we have are expensive, and resistance is developing to those drugs as well. They’re still our first line for preventing malaria in travelers, but not having a vaccine is a really important problem, and it’s something that we’re really working hard on-we and other people around the world.
So this resistance really meant that the malaria eradication program was derailed and that it was unlikely that Chloroquine was going to play an important role in eradicating malaria.
Interestingly, in 1962, President Kennedy in his inaugural speech talked about, “Let us eradicate disease together,” and he was speaking predominantly about the WHO efforts to eradicate malaria at the time. And in that same speech, he talked about taking man to the moon by the end of the 1960s, and we put humans on the moon in 1969, but we still haven’t eradicated malaria or even developed an effective vaccine against malaria now, nearly 40 years later.
How did resistance finally get to Africa?
We knew that Chloroquine resistance had developed as the parasite itself had become resistant. We’re not really sure how it came to be in sub-Saharan in Africa. It may have been that the parasite had mutated sufficiently to avoid being killed by the drug Chloroquine independently in Africa, or it could have been brought from somewhere else. But in any case, it developed initially in this one focus and then spread rapidly through sub-Saharan Africa.
What is the history of malaria as a disease?
We know that malaria was endemic in the American East, Southeast and even in the Washington, D.C. area during the Civil War, for example. There were a lot of casualties in the Civil War due to malaria; predominantly they were due to a species known as Plasmodium vivax. And malaria existed in the U.S. until probably the 1930s. I think most people attribute the elimination of malaria from the U.S. to the actions of Public Works departments and cleaning up the water.
Does malaria date back to the American Revolution?
During the Civil War, living and serving in the Washington, D.C. area I think was considered hardship duty, because of the hot, humid conditions that favored malaria transmission, and in fact, a lot of the people living in the area came down with malaria.
At the time of the American Revolution, we really didn’t understand what malaria was. I mean the parasite hadn’t yet been identified, we didn’t know much of anything about it.
When did malaria shift into developed countries, in modern terms?
Whenever business travelers and travelers from the developed world traveled into developing nations, they put themselves at risk for getting infectious diseases, and I think probably because of the increase in the amount of travel that occurred in the 1970s and 1980s that we did see an increase in the number of travelers that came down with malaria, and they went to these areas of the world where malaria was endemic, they were bitten by mosquitoes, and sometimes they came home infected with malaria and didn’t develop symptoms until they actually got home.
What about the military personnel?
Military personnel are a unique situation. There are two bits of data that are really quite important here. The first is that in World War II there were about 12 million man-days lost due to malaria in the world, and in Vietnam there were about a million man-days lost due to malaria.
If you look at the battlefield commanders’ reports, many times they would talk about not wanting to deploy into certain areas in Vietnam, for example, because the risk of malaria was too great. And so troops would go in, come back ten days later, and a large percentage of the troops would be casualties in the hospital because of having gotten malaria.
This is at a time when people were taking drugs-it didn’t happen to be Chloroquine-but were taking their chemoprophylaxis, their pills, which were supposed to prevent them from getting malaria. So for the military, malaria is a very important infectious disease threat, and that’s why the military has invested in developing new anti-malarial drugs and new vaccines.
Did the military personnel bring the parasite back to the U.S. in their bodies?
They would bring it back in some cases, but it’s not a significant health risk for example, to the U.S. population. People bringing malaria parasites back is not a big problem, because most of the time they’re symptomatic, and so they know that they’re ill and they get treated.
There have been cases where there have been outbreaks of malaria in the U.S. For example, I think there was an outbreak in the late 1980’s I believe in Southern California, where a Boy Scout troop actually had several cases of malaria, and it’s likely that there were migrant farm workers who’d come up from Mexico. They come up probably harboring parasites in their blood, and the mosquito vectors are here, and so mosquitoes bit the workers, picked up the parasite and then transmitted it to these Boy Scout troops.
So there are cases intermittently of malaria in the U.S., and of course, there’s the recent story of an outbreak of malaria here in Virginia, where several individuals came down with malaria. And they all came down with malaria at some distance from the airport.
There is a syndrome described as “airport malaria,” where mosquitoes hitchhike onto the airplanes and they’re infected with malaria. They get off with the people and they infect local residents, and so there are occasionally individuals who have never traveled and who get bitten by infected mosquitoes that come from endemic countries, and then they get malaria.
The strain of parasite that we are using in the malaria genome project actually was originally obtained from an individual who lived near an airport in Amsterdam. And so this parasite clone happens to be from the strain called NF54-now it’s a clone called 3D7-which was actually originally derived from a case of airport malaria. Interesting.
How many strains of malaria are there?
There are four species of malaria: Plasmodium falciparum, Plasmodium vivax, Plasmodium malariae, and Plasmodium ovale. Of those four, the one that causes the most deaths in really almost all the fatal cases of malaria is Plasmodium falciparum. Plasmodium vivax occurs probably in equal frequency, and it’s a pretty nasty disease. I mean you feel really bad, but in general, people don’t die from it. So it’s Plasmodium falciparum that is the one that causes the vast majority of the mortality in people.
How is global warming affecting malaria?
I think that’s still a controversial question. People have done modeling, looking for where there are incremental increases in temperature, and this might allow, for example, the mosquito to spread its bounds and that might introduce malaria into areas in the world that once had malaria, for example, but now are devoid of malaria. I think those are still just models at this stage, and it’s hard for me to really comment on that.
What is the cycle of the parasite in mosquitoes?
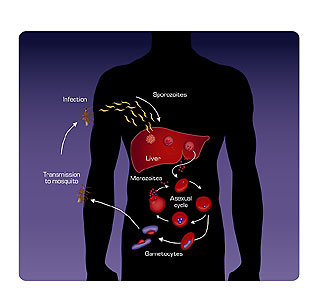
It’s a lifecycle, so you have to start at some place in the cycle. We’ll start with the mosquito, for example. So malaria-infected mosquitoes carry a form of the parasite called the “sporozoite,” and the sporozoite is found in the salivary glands of these mosquitoes. It’s a particular type of mosquito: it’s an anopheles mosquito. It’s one of the species of mosquitoes that carry malaria. These sporozoites are injected into people during the course of feeding on a blood meal by the female anopheles mosquito, which takes the blood meal so it’s the female anopheles mosquito that transmits malaria.
These parasite species actually get injected when the mosquito is feeding on the blood-the sporozoite form is released by the mosquito’s saliva during that process. It’s actually called Sporozoa at that stage. It’s called that because it is a discreet entity at that time. It expresses its own set of proteins that are different from the other stages of the parasite. The sporozoites enter into the bloodstream, and within really only a few minutes they are rapidly cleared, and they end up invading liver cells, individual liver cells. And over a course of about five days, they multiple to about 30,000 times their original number. These organisms are now called the merozoite form, which then burst out of the liver, and each of those individual merozoite forms invades a red blood cell.
Once they invade red blood cells, they begin feeding on the red cell’s hemoglobin-that’s the iron-carrying material that causes red blood cells to be red-and they feed on the hemoglobin and grow and divide to about 16 parasites in each cell. So they become packed with parasites. Those 16 parasites then burst out of the red blood cells. Each of those new forms invades a new red blood cell, and then they multiply 16-24 times, and then it bursts. So this happens about every two days.
All of the manifestations of disease really occur at that stage of the parasite. There’s no evidence of disease that is discernable when the parasite is still in the liver. It’s only once the parasite gets out of the liver that there’s any manifestation of disease, and this is where the fevers and chills and sweats come from.
Does this damage the liver?
They are cleared out of the bloodstream, and they get into the liver cells. It’s likely that they damage the individual liver cells that they invade, but there doesn’t seem to be any discernible pathology that occurs in the liver. And the numbers of sporozoites, which are being injected by the mosquito, are relatively small compared to the number of cells in the liver.
So the parasite then goes through this replication cycle inside the red blood cells. Now some of the parasites, once they invade red blood cells they don’t go on to develop and multiply inside the red cells to these next merozoite forms. They stop in their development-they stop in their replication but they go on to develop into male and female forms. And these male and female forms are called gametocytes. These gametocytes are circulating in the bloodstream, and when a mosquito then feeds, it takes a blood meal, and when it takes a blood meal it takes some of these male and female forms into its stomach, where fertilization occurs. This new fertilized parasite-called a “zygote” at this stage-attaches itself to the inner wall of the mosquito mid-gut, the stomach, and then burrows its way through the mosquito’s mid-gut and forms a cyst on the outside where it now develops and multiplies thousands of times, making individual sporozoites. These sporozoites burst out of this little sack and migrate their way through the body cavity into the mosquito’s salivary glands where the mosquito can then transmit them on its next blood meal.
And the really important thing to understand here, I think, is that what we’re dealing with is a separate parasite, really, at each of these stages. The parasite coming out of the mosquito, the sporozoite form, is a different parasite in essence, it’s a different set of proteins expressed, than the form of the parasite in the bloodstream. And it’s a different parasite probably inside the liver cell, and it’s a different parasite when it’s a sexual form, and it’s a different parasite when it’s actually in the mosquito mid-gut, each expressing a different set of proteins.
So if you’re thinking about developing new drugs or vaccines, you really have to keep that in mind. This isn’t, for example, like a simple virus or even a bacteria, which has really a limited repertoire of proteins that it can express. The malaria parasite has about 5,000 genes, probably at least 5,000 proteins, maybe more, that are expressed at different stages of the parasite’s development.
And this is one of the main reasons why we embarked upon the malaria genome project was to, first of all, catalog all of the genes that are present in the parasite’s DNA, and then to embark on a second project really, which was incredibly exciting, and I think probably has now raised the bar as to what is going to be expected of new genome projects, and that is trying to understand what are the proteins that are expressed in each of these different stages.
That way, we can have a better understanding about characterizing that parasite at each of those different stages. What is it about the parasite proteins that are expressed in the sporozoite, and how are they different from those expressed in the red cell stage, for example, and then can that give us clues in terms of what antigens we might be targeting for vaccine development and what drugs we might be looking at as well?
What amount of genetic material does the parasite have compared to a virus or a bacterium?
The malaria parasite genome has about 24 million based pairs of information in its DNA. And if we compare that to a typical bacterium, something like Haemophilus influenza, which has 1.6 million base pairs, you can do the math but it’s something along lines of fifteen times more DNA code. A virus might contain something on the order of 10,000 base pairs of information in its genetic code.
And so as you go up higher in the food chain, in essence, the bacterium is more complicated than the virus, and the parasite is more complicated than the bacterium. And the parasite has evolved to be fundamentally different from the bacterium. For example, the parasite actually has co-evolved with its host. It has developed adaptive strategies so that it can live with the host and survive really a huge onslaught of immune mechanisms that the host is sending its way to destroy it.
Does the vector, the mosquito, develop resistance to the parasite?
I think the mosquito has very limited repertoire-I’ll probably be chastised by the mosquito community here-but certainly the mosquito doesn’t have an adaptive immune system like we do. It has innate immune responses, and certainly it can evolve, but not the to extent that we can.
Has the mosquito evolved some resistance to DDT?
It has, but that’s really different. The mosquito has an innate immune system. It can’t respond rapidly to new infectious disease threats or to new pathogens like we can. We can develop antibodies and T-cell immunity. We can develop an immune response against a new organism we’ve never seen before. Mosquitoes can’t do that. Mosquitoes can evolve as a species over time. Their prodigy, for example, might be better able to survive carrying the malaria than its parents were, but as an individual, it can’t respond to a new infectious disease threat the way that we can. So the parasite and the mosquito have co-evolved much in the same way we have co-evolved with parasites.
A great example of this actually is the example of sickle-cell anemia. You know there’s a single nucleotide change, or mutation, in the DNA of individuals who have the sickle-cell trait. This mutation has conferred, to a large degree, protection against malarial infection in Africa, and if you look at where sickle-cell trait occurs and where malaria is found, the areas overlap substantially.
Now, this is an interesting situation for an adaptive mechanism. This trait should have been eliminated from the population. Right? If this mutation and this pathology had occurred without malaria present, it should have gone away; people would not have passed this on to their offspring because it could represent a lethal situation for individuals.
But because this sickle cell trait, this single point mutation, helped people to survive malaria, it persisted in the population, and so it’s a very interesting situation.
Can’t some people, if they’re chronically bitten by mosquitoes, develop some kind of immunity?
That’s really fundamental to our vaccine strategies. What I’m going to talk about is how we’re approaching developing malaria vaccines. We are focusing on two models. One is naturally acquired immunity, exactly what you’re talking about. How is it that people have developed immunity that allows them, once they reach the age of 10, basically to become chronically infected but not die or have a severe disease? Up to the age of 10 or thereabouts, there’s a significant amount of morbidity and mortality associated with malaria infection. The second model is something called “eradicated sporozoite model” which we’ll talk about in more detail in a moment.
How do you start to design a vaccine for malaria?
The first question really is, “What makes us think that we even have a chance in hell of making a malaria vaccine?” And, you know, vaccines that we have all taken, for example, a measles vaccine, I think that there was probably good evidence that a measles vaccine was going to be developed, and it was going to be effective, because if you get measles and you survive, you’re immune generally for the rest of your life. And so if one could replicate that type of immunity seen with the measles in a vaccine, you have a good chance that you’re going to be protected.
So we know that in nature, people who are infected with measles and survive are then protected. In malaria, that’s not the case. You can get malaria over and over and over again, and so if in early childhood particularly there’s a great chance that, even if you have malaria once, you’ll get it again, you might die. If you don’t die that time, maybe you die the third time you get malaria, or maybe the fourth time.
And kids who grow up in malaria endemic areas get malaria many, many times, and we don’t know exactly what occasion results in death in children, but certainly it’s not necessarily the first infection that kills children.
Are they being infected by different strains?
We don’t really know whether it’s a host factor, whether it’s in an individual’s genetic makeup that predisposes them to develop severe disease, or whether they happen to get, for example, an unusually heavy burden of malaria parasites, or maybe it’s a particular strain or clone that’s been transmitted. We’re not really sure.
There are studies going on actually in our laboratory in association with our colleagues in Ghana to look at this issue. It’s called a “case-control study.” We’re actually looking at severe disease, and we’re looking at not only what’s going on in people, but we’re looking at what’s going on in the parasite, and we’re using some fairly high-tech novel ways of looking at parasite diversity in those individuals. We’re trying to characterize them. Who are these parasites that are killing kids? Is there something unique about them as an organism, as a group, or is it something in individuals that is a factor?
And there are other things that are difficult to think about, like nutritional status and hydration status and other simultaneously occurring illnesses. They might have pneumonias. They might have diarrhea. There might be other factors, all of which can predispose them. So we’re looking at a lot of these things in studies like this case-control study to help us figure out what it is that predisposes kids to die from malaria.
Can we develop a cellular immunity?
So why is it that we think we can develop an effective malaria vaccine? Well, there are two models that we generally tend to use when we think of malaria vaccine development.
The first model is that of naturally acquired immunity. We know that kids who grow up in malaria endemic areas if they make it to the age of about 10 years of age, generally don’t after that point develop severe disease or die.
These kids, if you were to go into an endemic area in Africa, for example, and take blood samples from 12-year-olds or 15-year-olds who are apparently healthy, many of them have malarial parasites in their blood, but they’re not generally sick. And if they are sick, the cases are generally mild.
So what happens, we think, is that these kids are chronically and persistently infected with malaria, they go through multiple bouts of getting malaria parasites. And their body develops an immune response against the parasites. And we believe the predominant mechanism is against the parasite proteins expressed not only by the parasites themselves, but also the parasite proteins that are expressed in the red blood cells and on the surface of the red blood cells. And we believe that is the predominant immunological mechanism-an antibody response.
So people develop antibodies against parasite proteins expressed by the parasites, and they also develop antibodies against the parasite proteins on the surface of the red blood cell. It’s very interesting that the parasite, once it invades the red blood cell, tends to export its parasite proteins onto the surface of the red blood cell, and then sticks them into the surface of the red blood cell. So it actually alters the red blood cell itself, and puts parasite proteins onto its surface.
Are the proteins antigens that we react to?
An antigen is always a protein. We call it an antigen when it has relevance to the immunological system. So not all proteins are antigens, but all antigens are proteins.
So are the proteins that the parasites are producing acting as antigens?
They’re antigens if they become interesting to people developing vaccines. They become interesting to us who are looking at the immune response to those proteins. So we generally tend to focus only on using the term antigen when specifically speaking about a vaccine target, so not all proteins expressed on the surface of the red cell or expressed by the parasite itself may be relevant antigens. We just don’t know, and we’re evaluating that through some of these newer methods, these genomic methods.
If you’re developing a vaccine, don’t you have to know what protein the body is reacting against so it can produce an antibody?
The first model I talked about is the model of naturally acquired immunity. The second model is a more artificial model, and that’s the model of “irradiated sporozoite vaccine.”
We take parasite-infected mosquitoes, so these mosquitoes have malaria parasites in their salivary glands, and we subject those mosquitoes to gamma radiation-something like 15,000 rads of radiation-and we let those mosquitoes feed on people. We let 200 mosquitoes feed on a new individual for a ten-minute session once a month for six months. We then take the mosquitoes, which have malaria-infected parasites in them, allow those mosquitoes to feed on people, and the people who received these irradiated sporozoites are protected, sterilely protected, against malaria. 95% of the people who receive this “vaccine” are protected against malaria.
What we believe happens here is that the irradiated parasite gets into the blood stream just as it normally does, it gets to the liver, but because it was irradiated, it doesn’t go on to develop further in the liver, so it arrests its development inside the liver. So this gives the body an opportunity to mount a very strong immune response against the parasites and its proteins, the antigens, in the liver.
And we believe the predominant mechanism of that protection is a T-cell-mediated immunity, so a cellular-based immunity against the parasite proteins that are expressed in the infected livers, or in the specialized antigen-presenting cells in association with the livers, and if these parasite proteins are the ones that are being presented to the immune system, then it’s the cell-mediated immunity that is conferring protection in people.
And I’ve been through that vaccination process, by the way, and the problem is, it’s not terribly practical to immunize millions of people by allowing them to take a Dixie cup full of 200 mosquitoes with a mesh over it and put your arm in this cup and do this once a month for six months. It’s just not very practical. If it were working and were made more practical, it could be a fantastic vaccine.
So we have those two models. We have naturally acquired immunity, which we believe is predominantly focused on the blood stage, and we believe it’s predominantly an antibody-mediated immune response against the parasite proteins expressed by the parasite and those expressed on the surface of the infected red cells. That’s one.
The second model is that of the irradiated sporozoite. In this case, this is a cellular-based immunity against the parasite proteins expressed in the infected sporozoite. So using those two models, we think we can develop a vaccine, and we use those two models as we think about malaria development.
There is a program that has been focusing over the past ten years or so on some newer vaccine technologies, which we think give us the best chance of generating those complex immune responses. We need both antibodies and we need T-cell-mediated immunity, and we need immunity against several stages of the parasite simultaneously, and because it’s such a complex organism we probably need, again, immunity or a vaccine that attacks several different antigens on each of the different stages as well.
Did you purposely choose to attack the parasite in the liver form?
We’ve been approaching this with a vaccine that addresses both components. So our vaccine is designed to attack the parasite in the liver, generating T-cell mediated immunity against the parasite proteins expressed in the liver. We’re also addressing the vaccine by developing a vaccine capable of generating antibody responses against the parasite proteins expressed in the blood stage.
So the reason for doing that is that if we could completely protect people from the parasites getting out of the liver, we’d have a wonderful vaccine. It’s a hundred percent efficacious. It’s sterilely protecting, so the body would never see the parasites in the blood, and you’d never get any disease.
The problem is that if you have a 95% effective vaccine, 5% of the parasites may get out, and now because it’s actually a different stage of the parasite now you haven’t generated an immune response in individuals to combat that stage of the parasite. So the idea here is that we’re developing a vaccine that can attack the parasite in both stages, a pre-erythrocytic vaccine, which is the pre-blood-stage vaccine, as well as a vaccine that attacks the parasite in the blood stage itself.
Because this is a complex problem, we’re attacking the parasite at two stages. We’re asking the body to generate both a T-cell-mediated immune response as well as an antibody immune response, and we believe because it’s a complex organism, which would likely evolve around any of the particular vaccines we developed-against any one antigen-we think it’s important to target several parasite antigens simultaneously.
Most traditional vaccines don’t allow one to do that. And so we’ve adopted an approach, over about the past ten years or so, which focuses on DNA-based vaccines. DNA-based vaccines are more than just DNA alone. DNA-based vaccines include DNA, they include recombinant viruses such as recombinant pox viruses, recombinant adenoviruses, also something called “replicons,” as well as other technologies that are on the horizon.
So we’re evaluating those systematically, choosing a set of fairly well characterized antigens that many of us have been working on for a long time, putting these candidate antigens into these different vaccine delivery systems and exploiting our animal models to evaluate these technologies and try to get substantial protection in animals.
We’re actually very fortunate that we can use mouse malarias in mice, we can use monkey malarias in monkeys, and in addition we can use human malarias in humans. We have a clinical trial center where we can actually challenge individuals with the bites of infected mosquitoes and see if the vaccines work in people.
To a large degree, we’re in better shape than the HIV community, because they can’t do this obviously in people, and some of the animal models they use are less than adequate-their rhesus monkey system, for example. So we’re working heavily in the mouse malaria and in the monkey malaria and in humans, trying not only to identify new antigens, but also to develop new vaccine delivery systems that generate what we think is a required immune response against the selected antigens.
What is a four-gene DNA vaccine, and what are the four genes?
We have a large effort in our mouse malarias, and we’re focused in on a handful of genes in mouse malaria. Those antigens are the circumsporozoite protein. It’s a protein expressed by the sporozoite form of the parasite. We’re working on an antigen called SSP2, which is the sporozoite surface protein #2. We’re working on a protein called “HEP17,” which again is a mouse malaria antigen. We’re looking at an antigen called “AMA-1,” an apical membrane antigen. There’s one called “MSP-1,” which is a merozoite surface protein, as well as a few others in mice.
We’ve had quite a lot of experience in the mouse model, and over the past 18 months or so we’ve developed now a new model in rhesus monkeys. So we can take malaria infected mosquitoes-and these are Plasmodium knowlesi, which is a monkey malaria-we can take these mosquitoes, we can take them the sporozoites from them or allow the mosquitoes themselves to feed on monkeys, and we can challenge those monkeys to see if our vaccines protect against the sporozoite challenge, much in the same way that we would hope to be able to protect people.
In that model, we focused in on four antigens, the CSP, the Circumsporozoite Protein, SSP2, the sporozoite surface protein #2, AMA-1, apical membrane antigen 1, and MSP1-42, which is a fraction of the merozoite surface protein.
So these are four proteins?
Until the genome and the proteome project was finished, the way we thought these proteins were expressed was that the CSP and SSP2 were the pre-erythrocytic stage antigens, so these were proteins that were expressed by the sporozoite, and were likely to be expressed once the parasite entered into the liver. The other two proteins, AMA1 and MSP1-42, are parasite proteins, which are expressed predominantly in the blood stage.
Interestingly-and really this is only news that’s about a month old-we found that the AMA1 gene, and the AMA1 protein, is expressed not only in the blood stage but its also expressed in sporozoites. And this is information actually that came from the malaria genome project and from the proteome project, so it’s new information for us. So in fact our vaccine is two-stage exclusively pre-erythrocytic stage antigen, one antigen that is exclusively blood stage, and one that seems to have coverage in both.
In our human malaria vaccines, we’ve added an additional antigen to those, and that antigen is LSA1, liver stage antigen 1, and this is another protein that is expressed by the parasite inside the liver. Interestingly, we can’t find that similar gene in our mouse malaria models, for example. It may be that it’s in monkey malarias, but we don’t have enough information yet on that.
Is this a five-gene vaccine then?
The five-gene vaccine we’re working on is CSP, SSP2, LSA-1, AMA-1, and MSP1-42. Those are the five genes; it’s a five-gene vaccine that we’re working on for people.
Our group and others have been working on vaccinating by priming with DNA and then boosting with a recombinant pox virus or recombinant adenovirus or some combination of those four or five vaccine delivery technologies. The reason that we’re so encouraged in going forward with our human vaccine constructs to a large degree comes from our experience in our rhesus monkey model. Let me just tell you about this model.
Plasmodium knowlesi is a highly virulent and lethal infection in rhesus monkeys. Two sporozoites, two parasites that get into the bloodstream of a rhesus monkey will kill it. When we do our vaccine studies, we actually give a hundred sporozoites-50 times the lethal dose-and what we found is that when we do our vaccine studies, the control monkeys all rapidly get malaria, we give them drugs, and we cure them, so we’re able to cure the monkeys by giving them standard anti-malaria treatment.
In our vaccinated monkeys, very interestingly, for example in five monkeys, one would exhibit the same pattern as those control monkeys, so one rapidly gets malaria and needs to be treated. One of the monkeys doesn’t develop any malaria at all, it’s completely protected, so it’s gathering a substantial effect at the liver stage, certainly, and probably some effect at the blood stage as well. The other three monkeys from that group of five get malaria, but they don’t get a sufficient amount of malaria to put them in great jeopardy. They actually clear their parasitemia themselves. So what happens is, in those cases, we’ve given these monkeys sufficient immunity to allow them to clear the parasites from their own bloodstream. So really we’ve protected four out of five monkeys from lethal malaria with a combination of four-gene DNA vaccine prime recombinant pox virus boost.
Interestingly, that protection we’ve given them is substantial protection. If you were to re-expose them to more malaria, around that same time frame, they would probably clear the parasitemia much in the same way as they did the first time you got it. So you’ve given them sufficient immunity to allow them to clear their own parasitemia. But this particular vaccine doesn’t last very long, so if, in a couple of months, you were to go back and re-challenge those monkeys, the kind of immunity that we generated by that first vaccine doesn’t seem to persist, but it’s a first step for us.
I mean this is really one of the first times that we were able to demonstrate in this highly lethal malaria model that we were able to give substantial protection to these monkeys. And this is a highly lethal and virulent infection, probably much higher than people would get in the field, for example.
So based on that information, prime boost vaccines in our rhesus monkey work. We know that prime boost vaccines work in our mouse models. We are going forward, somewhat at risk, in developing these newer vaccine technologies, and it’s a long process. It might take a year or two years to get to the point where we can develop these vaccine technologies and put them into people.
Can you define what you mean by prime boost?
In this case, in this one example I told you about in the rhesus monkeys, what we did was we gave them DNA plasmids that encode each of four different plasmids, each of which encodes one of the antigens I told you about, and they’re mixed into a syringe and given into the muscle. And those monkeys were given three doses of that about one month apart. This is a bacterial plasmid. So that’s given about once a month for three months, and then we wait a little while, a couple of months. Then we give the monkeys a pox virus, and this is very similar to the smallpox vaccine actually.
Each of these pox viruses-there are four different pox viruses-each pox virus contains the same antigens, but the genes are encoded into the pox virus genome. So these are pox viruses that contain inside their genome one of the malaria antigens, and those are also mixed into a syringe and given to the monkeys.
And so what we think happens in that case is that the DNA somehow gets into either muscle cells or gets into antigen-presenting cells. The bacterial plasmid has something called a “SMV” promoter on it, which is a nucleic acid sequence that the body recognizes and will then use the DNA plasmid as a genetic blueprint to make the malaria gene into the form of protein.
So the cells that take up these plasmids then actually express the foreign protein. It’s a foreign protein. The body views it as foreign, and it generates an immune response. The immune response it generates-this primary immune response-doesn’t seem to be very strong. And what happens, we have found, is that if you come back and then give the body a different type of vaccine that codes for those same proteins, but somehow presents it to the immune system in a slightly different way, that now the body reacts much more rigorously against those parasite proteins.
At what stage is your research for a human vaccine?
One of the great things in science, obviously, is that we try to look at other research organizations, other groups, and see if we can’t learn from advances that these other groups have made, and see how we can apply what they’ve learned to malaria vaccine development.
A great example of this is the work that’s going on in the HIV community. The HIV researchers and people working on the vaccines are trying to generate strong cDNA T-cell-mediated immune responses, and they’re trying to generate T-cell mediated immunity much in the same way that we are against the liver stage.
There’s a recent publication in January (2002) where the HIV researchers did a comparison of DNA priming, pox virus boosting vs. DNA priming, and adenovirus boosting. And what they showed was that the adenovirus boosting was much better at protecting monkeys infected with HIV, and also in limiting the amount of diseases associated with these infections.
And so what we did is then was to exploit that information, and we’ve developed now adenovirus vaccines for our monkey models, and we’re in the process of developing adenoviruses for human malaria-challenged experiments.
So we’re using the monkey model that we developed with the prime boost strategies, building on that with DNA priming pox virus boosting and DNA priming adenovirus boosting, and then the results of that research are going to contribute to our understanding of what’s going to happen in people. And so we’ll be doing vaccine studies in people using again this combination of prime boost, multi-antigen, and heterogonous DNA prime recombinant viral boost in people, and hopefully we’ll be able to protect a significant number of people and then take those vaccines forward into field studies.
Have you already started clinical trials with humans?
Because this is a new technology, the FDA of course and other regulatory agencies requested we go relatively slow with this project, and we work very closely, as any vaccine developer does, with the FDA, making sure that all of our vaccines are made using good manufacturing practices, and that all these studies are done with full consent and approval of the institutional review boards, and if these are done they’re safe and ethical. DNA vaccines are a relatively new technology, and in fact our group was I believe the first to publish on the completion of a single gene vaccine given to healthy humans.
The FDA has requested that we go look first at single gene vaccines in people, and then we build on that and then go into multi-gene vaccines. So our first two or three studies actually were in human volunteers using the single plasmid. That’s Plasmodium falciparum CSP, circumsporozoite protein.
We then did a clinical trial of a five-gene vaccine. In this case, these five genes were all thought to be from the liver stage. In all of our trials, the vaccines have been shown to be safe, and they are slightly immunogenic, but they’re not widely immunogenic, and they certainly are not generating the kind of immunity that we think we would need to protect people against malaria.
But our main objective was to make sure that the vaccines were safe. And we know from our animal studies that DNA alone probably isn’t going to be sufficient to provide significant protection in people, and that we’re going to need a boost component to this. So while we’re developing the DNA component and doing safety studies in humans, we’re developing the boost component as well.
Now the DNA component is ahead of us on the timelines, so we have now a DNA vaccine that has been optimized in such a way that the DNA has been designed to more closely approximate that of humans, and the reason for doing that is that we’re asking the human cells to use this foreign DNA to make a foreign protein.
Well, the malaria DNA is very, very different from human DNA, and so what we found, and other people have found, is that if you synthesize the DNA so that it actually looks more like human DNA that the body then can recognize, it can make it into protein more efficiently.
Here’s an analogy of the problem with making foreign DNA into a human protein. Let’s say, for example, you are a car manufacturer. You’ve got an assembly line of people making Toyotas or Fords, for example. You have a foreman at the front who is reading the blueprints, and they’re all in English. Ford’s making great cars, and they can crank them out fast. If suddenly the foreman is now reading Japanese blueprints, production slows down, because the foreman can’t read the instructions appropriately to tell the workers how to make the cars. Okay? It’s the same machinery, it’s the same factory, but the foreman can’t read the instructions appropriately.
So if you think about it, malaria genes are so different from human genes that in essence it’s like a foreign language to the body, to the cells, and so what we did was we translated those foreign blueprints and we made them to read more like English, so that when the genes got into the human cells that the body could then use those blueprints more efficiently.
We know that immunization with DNA is inherently an inefficient process. Very little DNA actually is taken up by the cells, and so what we’ve done is to try to engineer our DNA to make it more efficient. We know it’s inefficient, that the uptake of the DNA into these antigen-presenting cells is an inefficient process. If it does get in, let’s make it as efficient as possible. So what we’ve done is, we’ve changed the genetic code to be more like that of humans so that the DNA, when it gets into the cells, is used most efficiently. So it’s making foreign protein as efficiently as possible, and so it’s producing the foreign material, and then the body then has more foreign material to react.
Do you have the whole genome sequence for the mosquito, the parasite and the human?
In 1995, Dr. J. Craig Venter published the first complete sequence of a free living organism, that of Haemophilus influenza, and he published that in Science, and immediately Dr. Steve Hoffman, who was the director of the malaria program at that time, recognized this as being potentially a really important tool in the development of new vaccines and new drugs against malaria-this idea of determining whole genome sequencing.
I had just arrived actually to the malaria program here, I hadn’t done my PhD yet, and he and I and others actually met during the blizzard of ’96, pushing our cars up the ramps of Highway 270, and we met at The Institute for Genomic Research to talk about how we would go about sequencing the malaria genome.
In parallel, a group at the Sanger Centre in the U.K. was also thinking about doing malaria genome sequencing, and over the next several months it became clear that an international consortium needed to be formed to address this major problem and also to get sufficient funding to carry the program forward.
So this was an international consortium of funders from the Department of Defense, from the Burroughs Wellcome Fund, from the NIH, and from the Wellcome Trust in the U.K., which funded basically three major genome centers, the Institute for Genomic Research and the Naval Medical Research Center working together, the Sanger Centre in the U.K., and Stanford University as the three major genome centers working together on sequencing the malaria genome.
There was a lot of skepticism amongst the malaria research community and others that this could be done, and that’s because the malaria genome is so different from any other genomes that have been sequenced. The genetic code is made up of four different nucleotides of course, the letters A, C, G, and T. And in humans and in most mammals and most organisms, it’s quite a nice balance between A, C, G’s and T’s in the DNA.
But in malaria, the Plasmodium falciparum, the organism that we were sequencing is very much biased towards A and T. So it’s about 80% A’s and T’s, and in some regions it’s almost a hundred percent A’s and T’s.
And so, for some technical reasons that are difficult to explain, many people thought that it wouldn’t be possible for us to chop up the DNA, sequence it, and put it all back together correctly. But we were able to do that. We took a million base pairs from Chromosome 2, and really in only about 18 months we were able to complete the entire sequence of Chromosome 2, and we published that in Science in 1998. So really we established in principle that the genome project would be successful, and then there was a major effort at that point to ramp up the sequencing and to get the whole Plasmodium falciparum sequence completed, and then of course you know we published that sequence in the October 3rd (2002) issue of Nature.
But in addition, because we learned so much about doing sequencing with malaria, we decided to tackle one of the model parasites that we use for vaccine development and some people use for drug development, called Plasmodium berghei-it’s a mouse malaria-and we sequenced that rapidly, and really in less than 18 months we completed the entire sequence of this mouse malaria, and the interesting thing was that it allowed us to make comparisons of two different species, in essence at the same time. This usually doesn’t happen. Usually you do a sequence, and someone does another sequence; another organism may be sequenced by another group, and eventually after a little bit of time some comparisons are made.
So I think really this is a very unique opportunity: to do the sequencing of two organisms simultaneously, and to compare the genomes, to look at what are the differences and similarities between these two really quite different species.
And at the same time, about two years ago actually, I was visiting Washington State University, and I heard a talk by a guy whose name is John Yates, who was working on proteomics; he worked with yeast. And developed an approach called MUDPIT, Multi-Dimensional Protein Identification Technology, as a means for identifying the proteins that are expressed in yeast. And it struck me that the kinds of work that he’s doing and the approach that he’s taking to yeast are immediately applicable to the problem we had with malaria, which is trying to determine which proteins are expressed at the different stage of the parasite’s lifecycle.
We embarked on a collaboration together to actually do the entire proteome analysis of four different stages of the parasite simultaneously. And so in the same issue of Nature, we had the whole genome of Plasmodium falciparum, the whole genome of Plasmodium malariae, and the entire proteome from four different stages of the parasite.
And I think what that has done is to really raise the bar as to what’s I think is going to be required from future genome projects, but it also has sparked entire new areas of malaria research.
For example, we found that the parasites in the sexual stage before it gets the mosquito use a completely different energy metabolism system than the blood stage parasites, and that has implications for drug development, for example.
There is one parasite protein that we thought was exclusively involved in the blood stage, which we thought was really important for antigenic variation, for avoiding the parasitic immune system, and possibly for what we see in clinical disease, which is the attachment of the parasite to the inner portions of endothelial cells.
What we found was that these sets of proteins, which were thought to be exclusively in the blood stage parasite, are actually highly abundant and they’re expressed in a very diverse way in the sporozoite form of the parasite, and this was something that no one had ever demonstrated before.
Aren’t they like entirely different organisms in different stages?
Determining the proteome really gets at this question, which is trying to figure out what it is about the parasite in each of its different stages and why has it evolved the way-it has to have many different parasite stages to have different more morphological forms, in order to specifically invade liver cells, to specifically invade red blood cells, and not potentially invade other cells, to go on to develop into the sexual forms, and so on. It’s really incredibly fascinating. But yes, it is like a different organism at each stage, and knowing the genome really doesn’t help you in that area, because the genome’s the same in each stage. The genome in the sporozoite is the same as in the merozoite, and as the gametocyte. It’s all the same. What’s different is the gene expression and really probably more importantly, the proteome, or the proteins that are expressed at each of those stages.
And the proteome project really gets at that. It addresses what parasite proteins are expressed at those different stages, and not only that but where they’re expressed. Are they expressed in the surface of the parasite? Are they expressed inside the parasite?
How will that help you in development of a vaccine?
We are working with antigens that were discovered probably over 15 years ago, and we are moving forward with those vaccine candidates. They’re very well studied. They do represent parasite proteins, which are expressed at different stages of the parasite. We really don’t know at this stage whether they are the best ones to use in a vaccine.
And the problem is that there isn’t a really good method that anyone has developed to help us go from the genome or the proteome to determine whether any antigen is better than another one, or which antigen is the best one to pick from.
Part of the work here at the Malaria Program at the Naval Research Center is actually to attempt to address that problem, and we’re using bioinformatics approaches, computer-based searches, we’re looking at the proteome in trying to select and to prioritize the parasite proteins expressed at the different stages’ proteomes. We’re using our animal models to help discern whether any of these antigens are better in our animal models, and we’re also exploiting the eradicated sporozoite volunteers, because we know they’re protected. So these individuals are protected, and we can look at their immune system and try to determine what is it about their immune system that’s responding to the parasite proteins expressed by the eradicated sporozoite, and can we identify those same proteins from live parasites.
Is there a different strategy for developing this vaccine compared to others because it’s such an ancient disease?
Here we are dealing with a disease that is really ancient, and it has co-evolved with us for many, many, many years, and I think because of the complex nature of the organism and the complex nature of the disease and the complex nature of the immune response that occurs in individuals, that we have to apply the best and the most up-to-date technologies to fight this disease. And those include genomics and proteomics, and it includes DNA-based vaccine technologies, as well as basic research and fundamental biological studies.
Our hope really is that the work that the Malaria Program is doing is going to benefit not only the development of vaccines against malaria, but through our collaborators, it will provide a better understanding of the malaria parasite overall.
Certainly the proteome has sparked a whole new area of research. One, we’re embarking on a collaboration, for example, to study host-parasite interactions inside the mosquito, trying to figure out how a mosquito responds to the parasite and how the parasite responds to the mosquito. Well, now that the whole genome of the parasite is completed and the whole genome of the mosquito is completed and we have tools such as proteomics available to us that we didn’t have a year ago to begin to address what’s going on in the parasite and what’s going on in the mosquito. It’s really very exciting.
And this has led you to think about transgenic mosquitoes that can’t carry the parasite?
Certainly as a model that really is a very interesting idea, because the transgenic mosquito model might tell us more about the biology. You know, the transgenic mosquitoes couldn’t be released; they might not be a control strategy that would be palpable to environmental groups, for example, or to scientists probably as well.
But what happens in those mosquitoes might give us a clue on how we could, for example, develop some other interventional strategy. We might be able to develop a vaccine, for example, which actually protected the mosquito from malaria.
For example, you could develop a vaccine that develops antigens that are taken up by the mosquito in a blood meal, and those antigens might block invasion of the parasite into the mosquito gut. And people have done this in the past. I mean this is not new news. I’m not bringing up anything that hasn’t been done in the past. People have looked at adding, for example, antigens into blood that’s infected with malaria, and people have shown that those antigens can actually block the invasion of the parasite into the mosquito stomach, for example.
Are you able to ask questions about the human, mosquito and the parasite are interacting all at the same time now?
I wouldn’t be so bold as to say we’re able to answer those questions now, but we’re thinking about it.
We’re certainly thinking about it, and I think that we now have technologies that we can begin to start looking at those host-parasite interactions. We know a little. Obviously, in any parasitic organism there is an intimacy between the parasite and its host, and I think there’s a lot that can be learned from what those interactions are. If we have a better understanding of how the parasite interacts with the hosts-human and mosquito-I think we’re going to have a leg up in terms of developing targeted interventional strategies against the parasites and trying to disrupt those interactions. It’s a big job.
What is the importance of continuing public health ventures along with research to help control malaria?
There’s a lot of effort going on in malaria vaccine development and drug development, but children are dying every minute, and so it’s imperative that public health measures are maintained and increased to help the children of today to make it to the age of 10 when they’ve developed naturally acquired immunity.
A vaccine is not around the corner. Even if we had an effective vaccine today, it’s still years before that vaccine would be able to complete the necessary clinical trials in support of licensure, and we don’t have a vaccine in hand right now that we believe is optimal and that is going to give us the kind of protection that we’re going to need, so we are years away.
Certainly, once we have substantial protection, and the kind of vaccine that is likely to make it all the way through the development process, then I think there’s hope out there, and then it’s just a matter of making sure that the vaccine continues to be safe in larger and larger numbers of people, and then its efficacy is maintained and that we haven’t developed a vaccine that only protects against the laboratory strain and doesn’t protect against all the natural strains.
What are some of these health measures that need to continue?
I think the kinds of public health measures we’re talking about, to a large degree, depend on the areas in which people are trying to control malaria. The kinds of interventional strategies that people obviously are working on are mosquito control, using nets and reducing the mosquito populations, good case control, looking at an adequate health care.
And some of those areas are difficult for many people, because they’re expensive, and third world developing nations healthcare budgets, if they were to attempt to implement all of these preventive measures, it would completely deplete them of all of their health dollars. So the kinds of interventions include bed nets, which have been shown to reduce childhood mortality by 30% in some areas, so they’ve really made a great stride.
What is the state of the research in humans with the five-gene vaccine?
We completed a Phase 1, 2A study with a five-gene vaccine. Those five genes were not optimized genes, and they were all directed against the liver stage or the pre-erythrocytic stage vaccine. In that study, that vaccine was shown to be safe, but it wasn’t very immunogenic and it didn’t protect people.
So what we’ve done now is gone and increased the number of antigens under our scrutiny, so we’ve gone from five to nine, and then we looked at those in our systems here, and now we have selected them down to a different set of five antigens. So the five antigens that we’re moving forward with in our current development strategy are three pre-erythrocytic stage vaccines, and so we’ve eliminated two from the original set of five and we’ve added two vaccines from the blood stage.
Are these being tested on monkeys now?
These vaccines currently are undergoing preclinical immunogenicity studies. This is a DNA vaccine cocktail that’s undergoing preclinical studies in mice and in rhesus monkeys, and depending on that immunogenicity, we will decide whether that or not they will go into clinical trials in people.
But again, I just want to stress that we view this as only the first component of a prime boost vaccine. We are now in the process of actually designing and constructing and manufacturing the recombinant boost components of these vaccines, which include both pox virus and adenovirus.
What are the big questions you’re still trying to answer?
Malaria kills people really by two methods. One, it causes severe anemia in those individuals infected, and we think there are two ways that this happens. One is that of course, when the parasite bursts out of the red cell, it destroys the red cell. So there is erythrocyte degradation-the red cells are being destroyed by the parasites themselves. The second way is that we know that the parasite, for some reason, also suppresses the body’s ability to make new red blood cells. It’s called erythropoiesis, and so there seems to be some suppressive effect actually at the bone marrow in producing new red blood cells, and we don’t really understand why that happens.
So one way that children die of malaria is through severe anemia. The second mechanism by which malaria kills people is through the development of cerebral malaria. And what happens in this case is that the parasite expresses on the surface of the invaded red cell parasite proteins, which for some reason interact with human proteins that are expressed on endothelial cells. And endothelial cells are those small vascular cells-the cells actually that are in the lining of the blood vessels. So the parasite sticks to these human proteins in the inside of the blood vessels, and they get plugged, so you actually have parasitized red cells that stick to the small vessels inside the brain and cause “sludging,” and you end up getting blocked capillaries and blood flow is reduced to those areas. Children develop seizures, coma, and eventually they die from cerebral malaria.
So there are two means by which people die of malaria, generally from severe anemia and from cerebral malaria.
When you look at malaria, what happens is that malaria is in many places in the world seasonal, so that as the rainy season comes malaria comes, and when the dry season comes malaria tends to go away. And what you can see very clearly is that, particularly in kids, that the amount of anemia that develops with the rainy season becomes extraordinarily high. They develop profound anemias, but in many cases they tend to recover their blood counts.
What are the big questions left?
As we approach malaria vaccine development, and as one approaches any vaccine program, you have to first of all try to identify the important vaccine antigens. Now, again, as I said, we inherited a set of antigens, and we’re moving forward with those. We’re hoping that the genome project and the proteome project is going to give us clues that we might be able to use to find better sets of antigens for our next generation of vaccines. So that’s one area.
The second area we think we understand quite well: how the body responds to naturally acquired immunity into the eradicated sporozoite. We think the body responds by generating antibody responses against parasite proteins expressed either in the blood stage or in the liver stage.
The question we have is, “what are the parasite proteins that the body is reacting to? Are the immunological responses that the body generates-are they protective? And how do we generate a vaccine delivery system that allows the body to generate the appropriate immune responses against those that affect parasite proteins?
And so that’s a challenge for us, and that’s really the foundation of the malaria program here is generating the appropriate immune responses against the parasite proteins that we think are important using the vaccine technologies that are conventionally to be put into people and licensed by the FDA and eventually distributed to the world.
Do you think your work with DNA-based vaccines is the strategy that will succeed?
There is substantial effort going on in recombinant protein-based vaccines, and I think that certainly the vaccine that’s the furthest along from field testing in fact is a recombinant protein vaccine-and I think recombinant protein vaccines are an important part of the vaccine development, and I think we need to continue to invest in the recombinant proteins. There’s a lot of history. We know a lot about how the body responds to the recombinant proteins.
The approach we’ve taken, and other people have taken, with our DNA-based vaccines really may be complementary to that. I mean it may be, for example, that one of our DNA-based vaccines might be a prime, and we might boost through a common protein, for example.
But I think that what we’re asking the body to do is to generate broad immune responses, both antibody and T-cell mediated responses against a whole series of antigens simultaneously. I think that it’s going to be difficult for recombinant proteins to be able to do that, and I think that the DNA-based vaccines are likely to provide the best method for being able to generate those kinds of immune responses.
If we had to generate only antibody responses, recombinant proteins would probably be adequate, but we’re asking the body to generate not only antibody responses but also cellular-based immunity, and recombinant proteins in general don’t generate good T-cell mediated immunity.
Could you give a definition of the comparison between DNA-based vaccines and recombinant proteins?
The fundamental difference between DNA-based vaccines and recombinant protein is that, in the case of DNA-based vaccines, you’re asking the body to produce the foreign protein using its own cellular machinery.
With recombinant protein vaccines, you use a bacterial system or a yeast system or some other insect cell system in vitro, in a laboratory, and you put the genetic code into those cells in a flask or into a bottle and you ask those cells in the laboratory to make the foreign protein. The foreign protein then has to be purified from all of the insect cells or the bacterial cells. It has to be purified and then it’s delivered generally with some sort of immune enhancer to a person.